The fifth version of ISPE’s The Good Automated Manufacturing Practice (GAMP 5) Guide for Validation of Automated Systems in Pharmaceutical Manufacture represents the most recent and updated tool in the approach for the validation of GxP computer systems. If computer systems employed in the pharmaceutical industry do not follow GAMP 5 guidelines, they have a risk of non-compliance with FDA’s directive 21 CFR Part 11.
Fifteen years ago, Able Laboratories was one of the top producers of generic drugs. But that was before FDA investigations. Following audits in the 2000s, the Food and Drug Administration discovered that the company had falsified documents in order to market potentially dangerous drugs. Today, Able Laboratories is history, and an example of what non-compliance with FDA regulations can lead to.
It was while ensuring compliance with FDA 21 CFR Part 11 that the FDA discovered irregularities. This regulation details all the procedures to follow regarding document security and electronic signatures. The objective is to avoid any falsification or attempt at concealment. By putting the finger on a bad document management, the FDA thus broke off the scandal and caused the fall of the big pharmaceutical giant.
If you want to avoid unnecessary inspection procedures and short audits, there is a simple rule: follow GAMP 5 guidelines.
GAMP 5 guidelines: a risk-based approach to FDA validation
GAMP version 5, based on the key requirements established by the FDA in 2008 proposes to validate, step by step, the compliance of document management systems. It is based on five key principles, namely:
- The understanding of the product and the process in relation to the IT system
- An approach based on a life cycle of the computerized system within the framework of a Quality Management System
- The scalability of life cycle activities
- Scientific (rational and documented) approach to risk management
- A valuation of the involvement of the system supplier
The major novelty of these GAMP 5 guidelines is its approach. Each compliance action must be defined with respect to the risks associated with the use of the system. What’s really interesting is that it requires very concrete questions to be asked from each of the computer tools and platforms involved, particularly for Learning Management Systems (LMS).
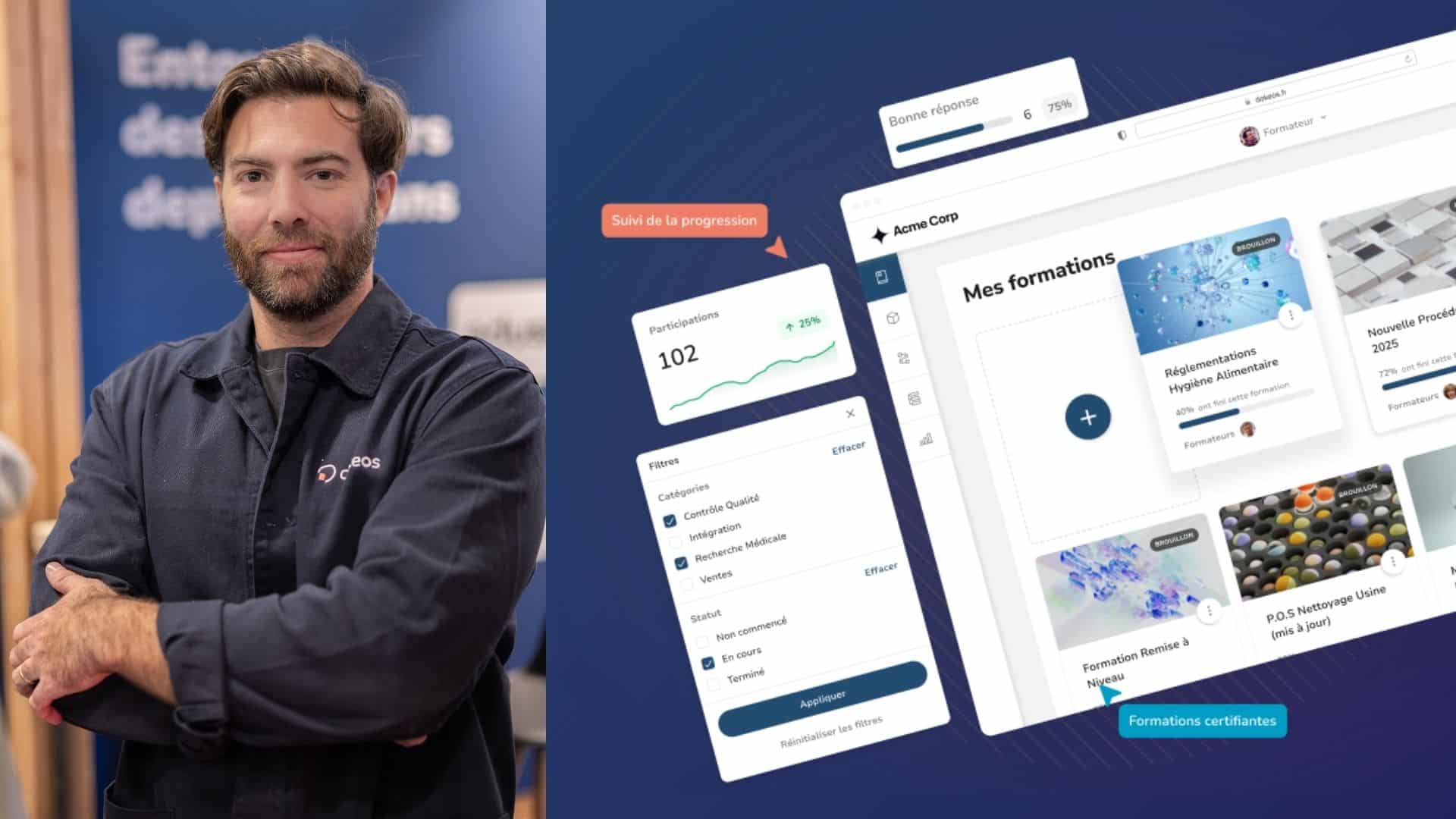
Dokeos LMS, in line with GAMP 5 guidelines
The purpose of an LMS in case of an FDA or EMA audit is to show that the laboratory, dispatcher or supplier has trained the right people, at the right time, in the right procedures. In short, that it is in line with its training obligations, without possible doubt nor falsification.
Precisely, these are the rules to be followed, and which Dokeos LMS includes by design:
- Each training course must be documented on paper, with a list of employees who completed the course and obtained a diploma or certification. This document must be reliable, authentic, dated and signed.
- Each employee must have his/her training file, with training courses followed in detail (a simple YES / NO is not enough), and diplomas and certificates attesting to skill acquisition. At each step, an electronic signature is required to guarantee the authenticity of the user.
- All SOPs (standard operating procedures) must be signes and approved by all employees involved in the process.
- There are also certain rules on the questionnaire side, with numbered and blocked multiple choice questions.
- Access control, with various attributions and rights according to the roles of each individual (trainee, trainer, manager, HR)
- Encryption and data protection to prevent forgery. The platform must, for example, prevent any file destruction or replacement, unless this replacement is traced, dated, and justified.
Contact us to discover all the features of Dokeos LMS